

Abdulaziz (Omar) Badran: Navigating the Clinical Complexity of Combined Red Cell Disorders
Abdulaziz (Omar) Badran, Hematology Supervisor at Almoosa Specialist Hospital, shared an interesting case on LinkedIn:
“Good evening to all.
Our case today considers rare even we have them from time to time in this area.
22 years male came to ER diagnosed in other hospital with sickle disease and he is blood transfusion dependent.
So he was admitted directly and had blood transfusion due to very low HGB 5.4 g/dL.
So directly they gave him blood and his HGB became 9.19 after double transfusion.
We can see in addition to mild microcytic hypochromic moderate anemia with high RDW.
High Retics 17.2 %. he also has mild Leukocytosis with Neutrophilia.
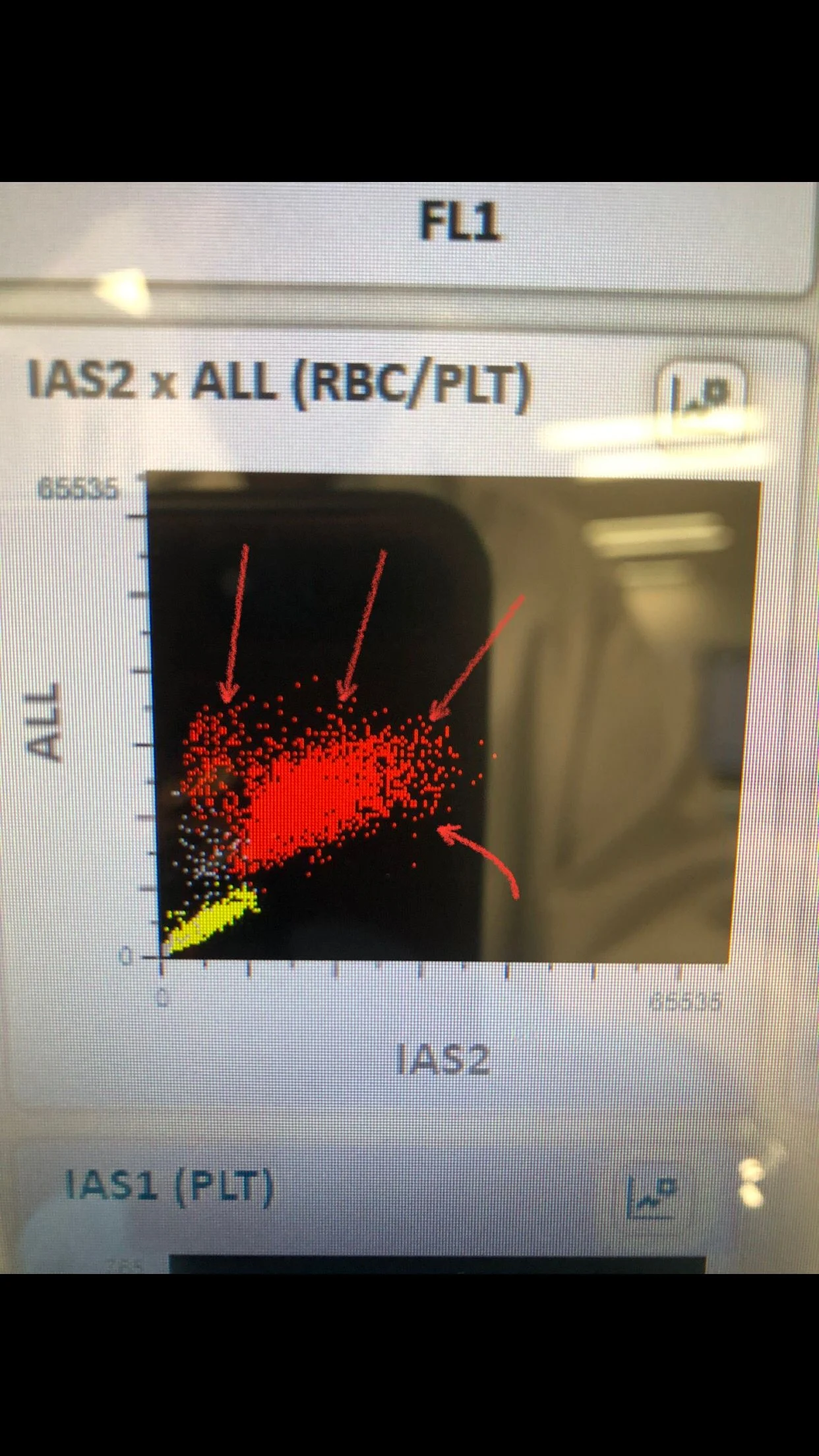
Also rstRBC flag which come usually with Schistocytes or sickle cells. and RBC fragments flag which is usually for Schistocytes or Spherocytes which can be interfere with platelets count.
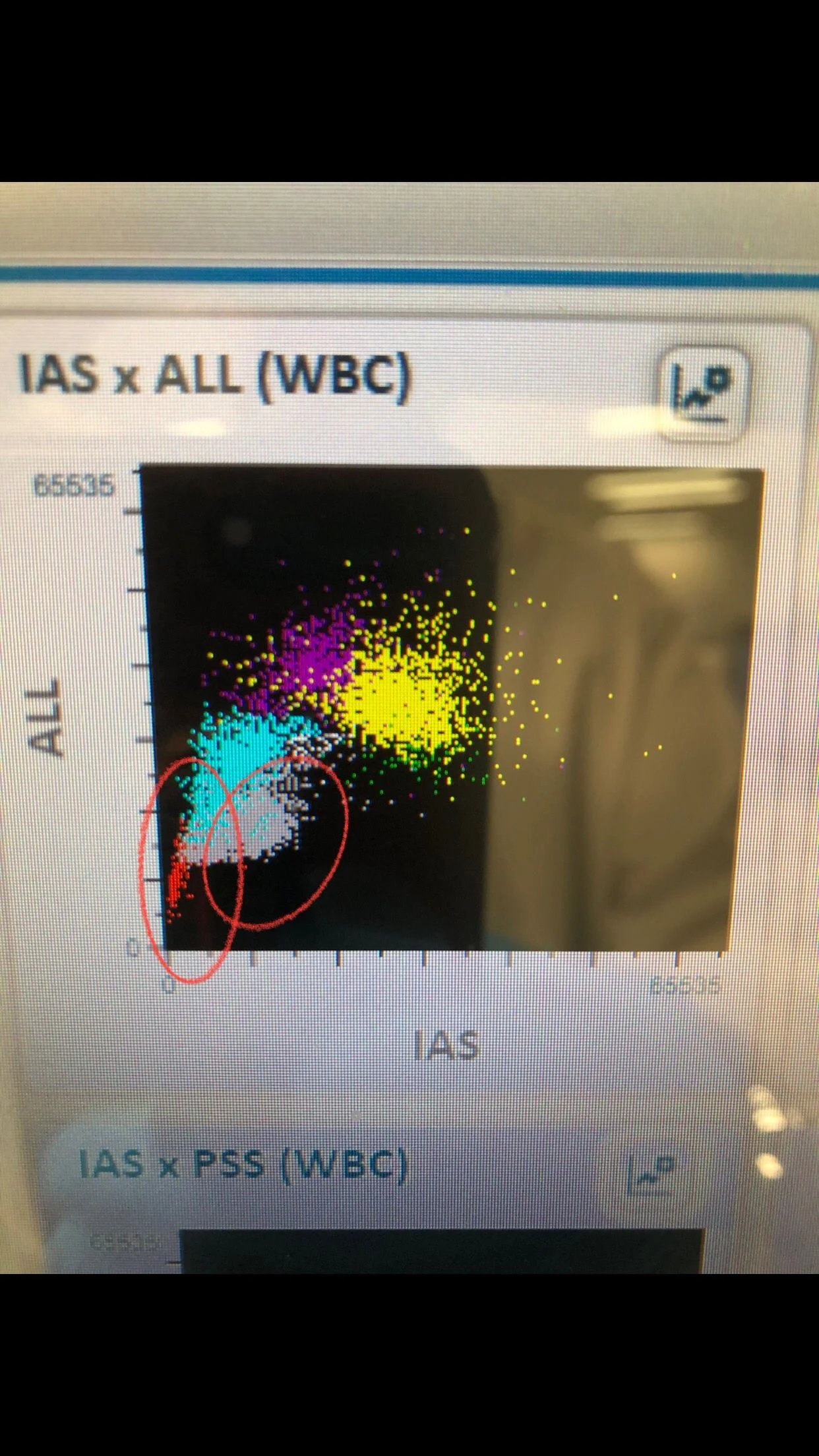
WBCs scatter shows majority of Neutrophils (yellow color), gray color scatter for Schistocytes or sickle cells (unknown remains cells not WBCs), and very few red color scatter for the few NRBCs present in the sample.
RBCs scatter shows 4 different populations and there is clear area between RBCs (red), and platelets (yellow) meaning the machine count them accurately and there is no interference between both.
The slide unfortunately was disaster.
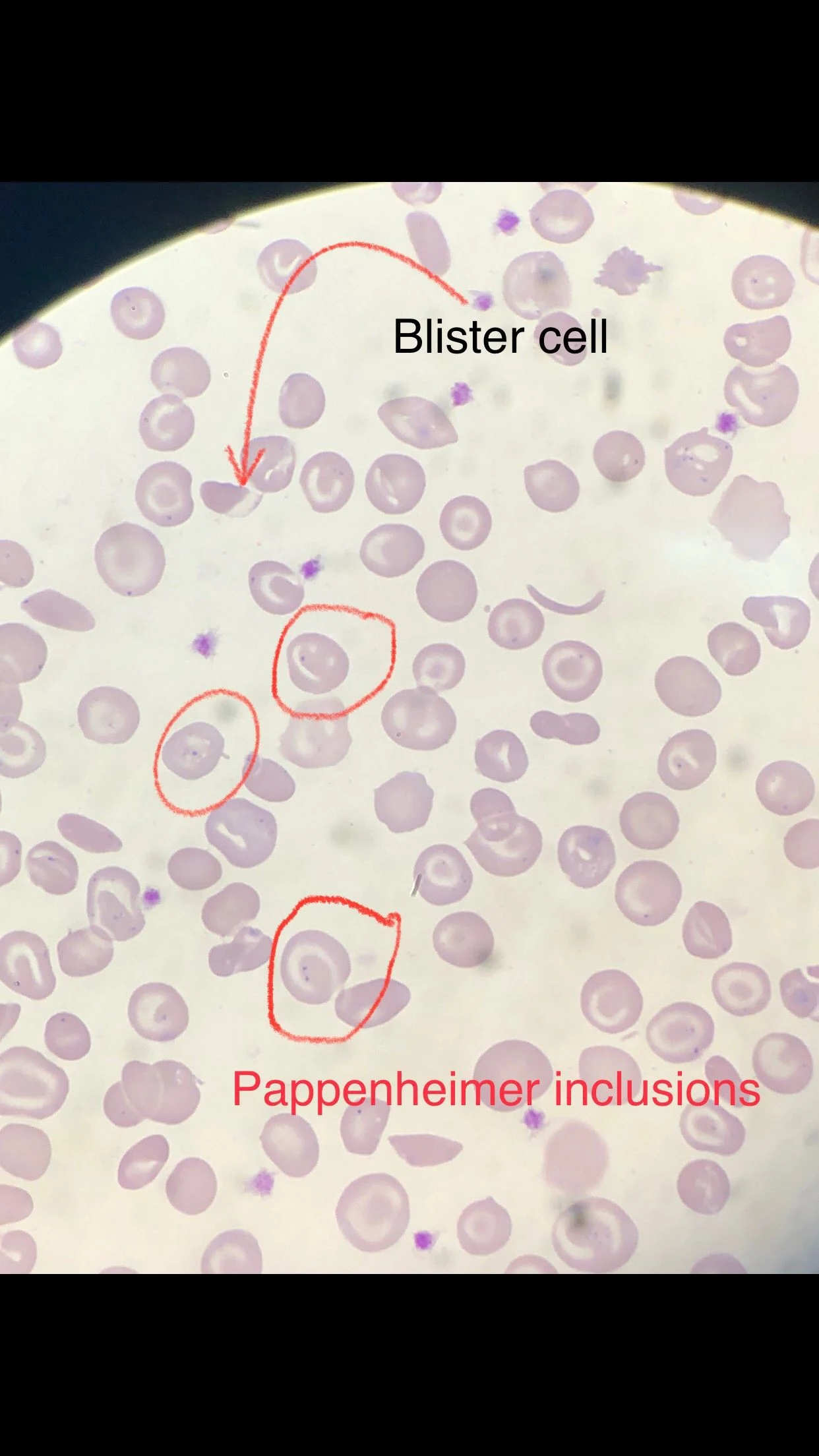
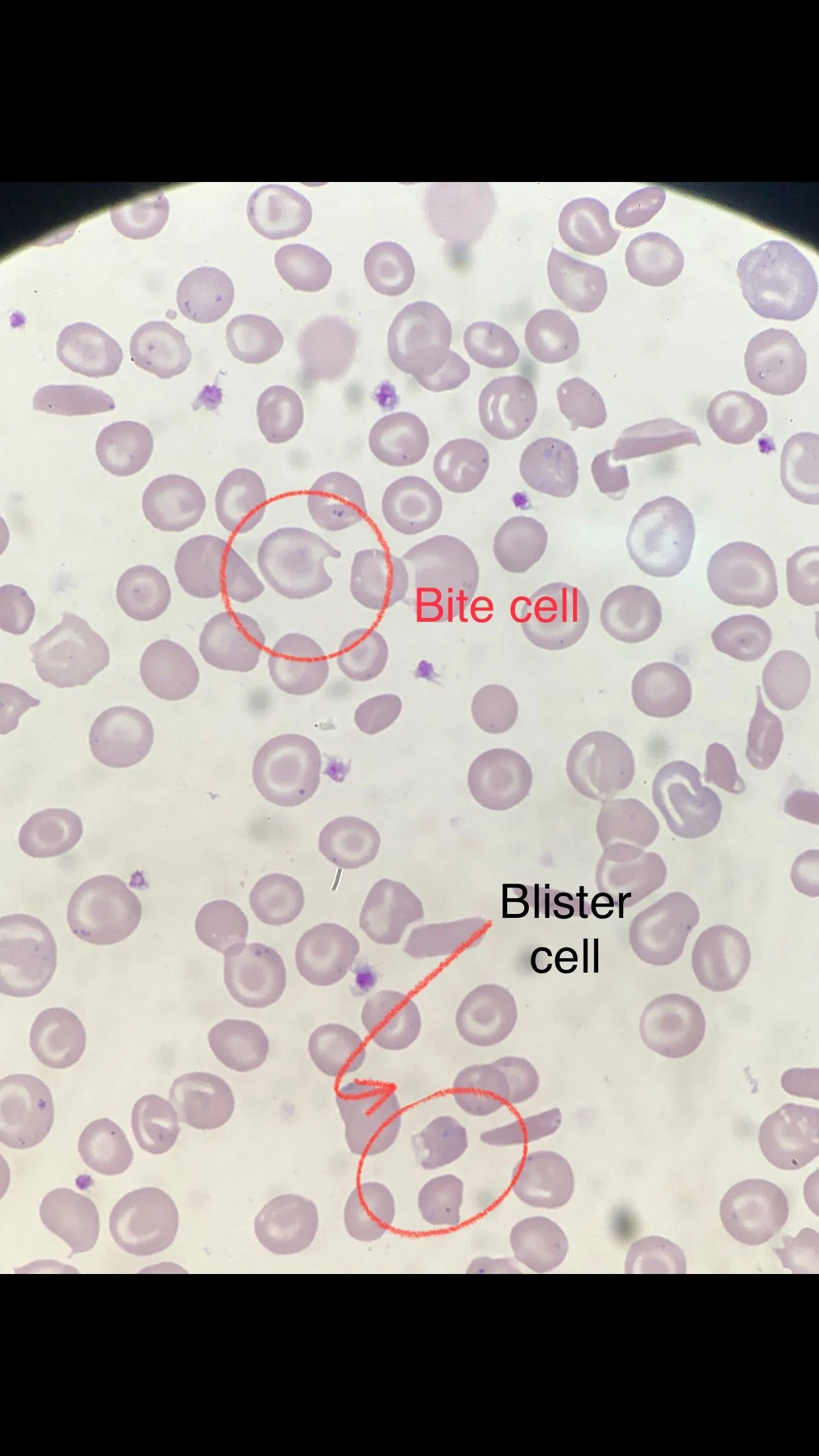
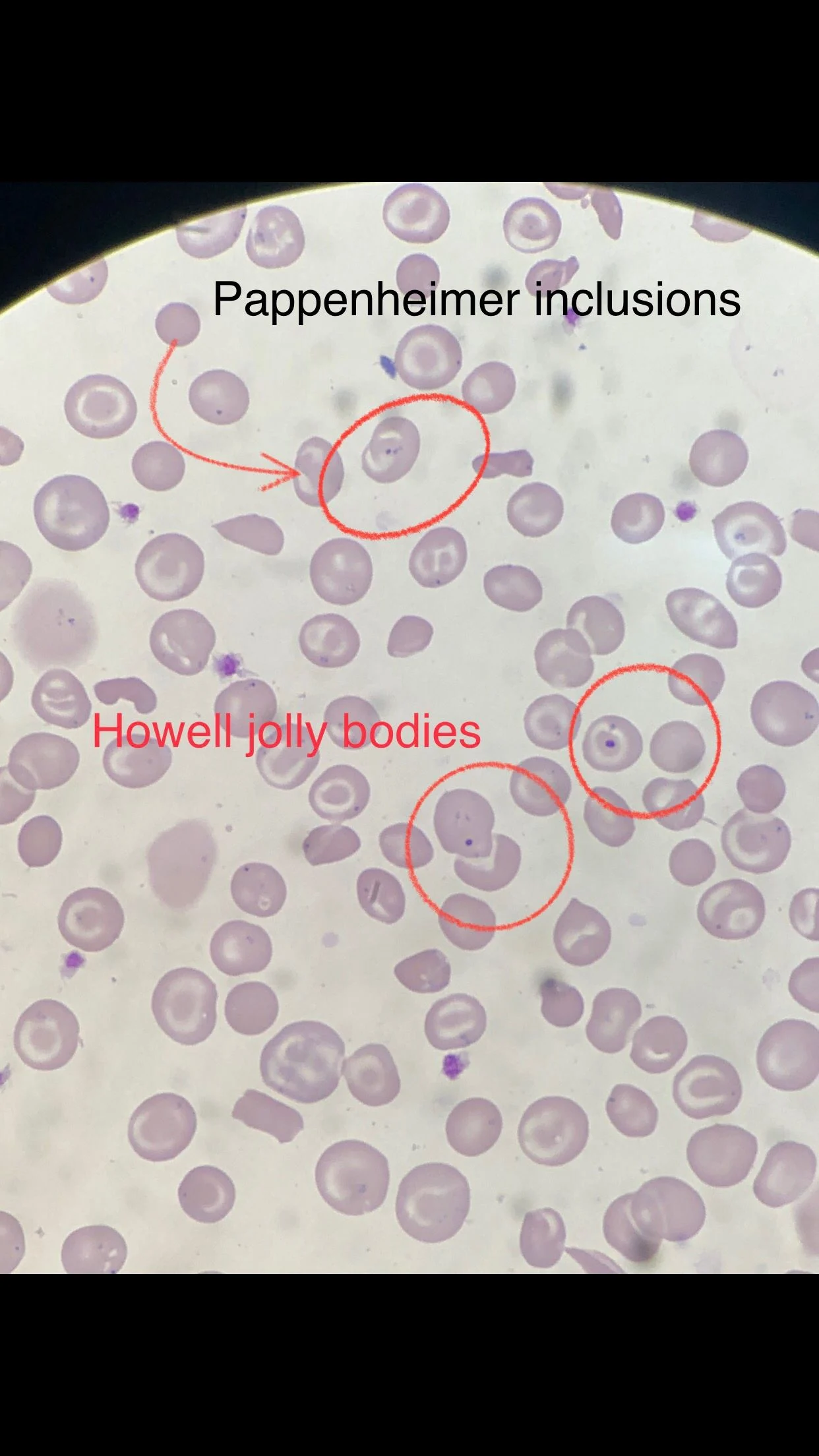
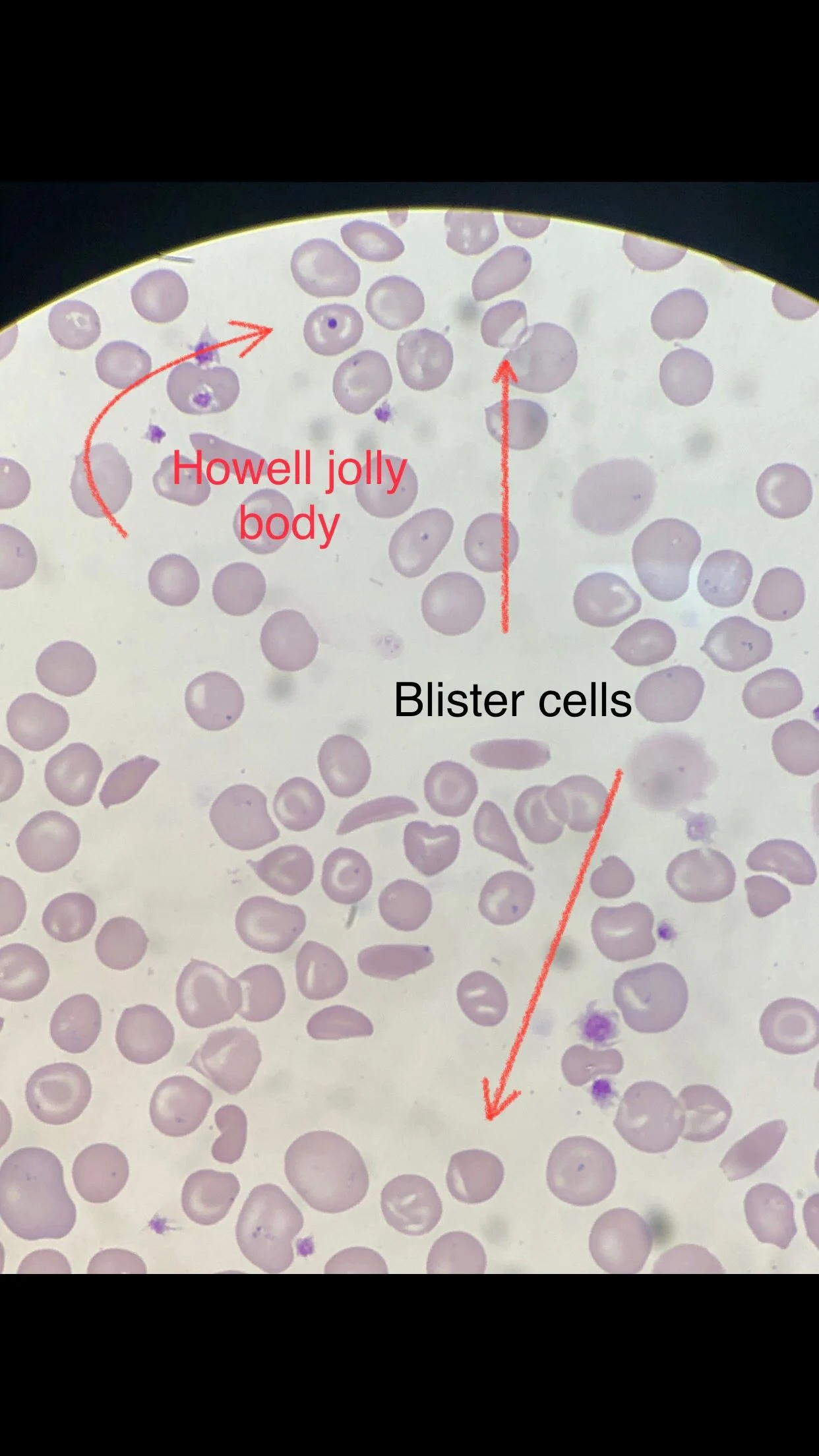
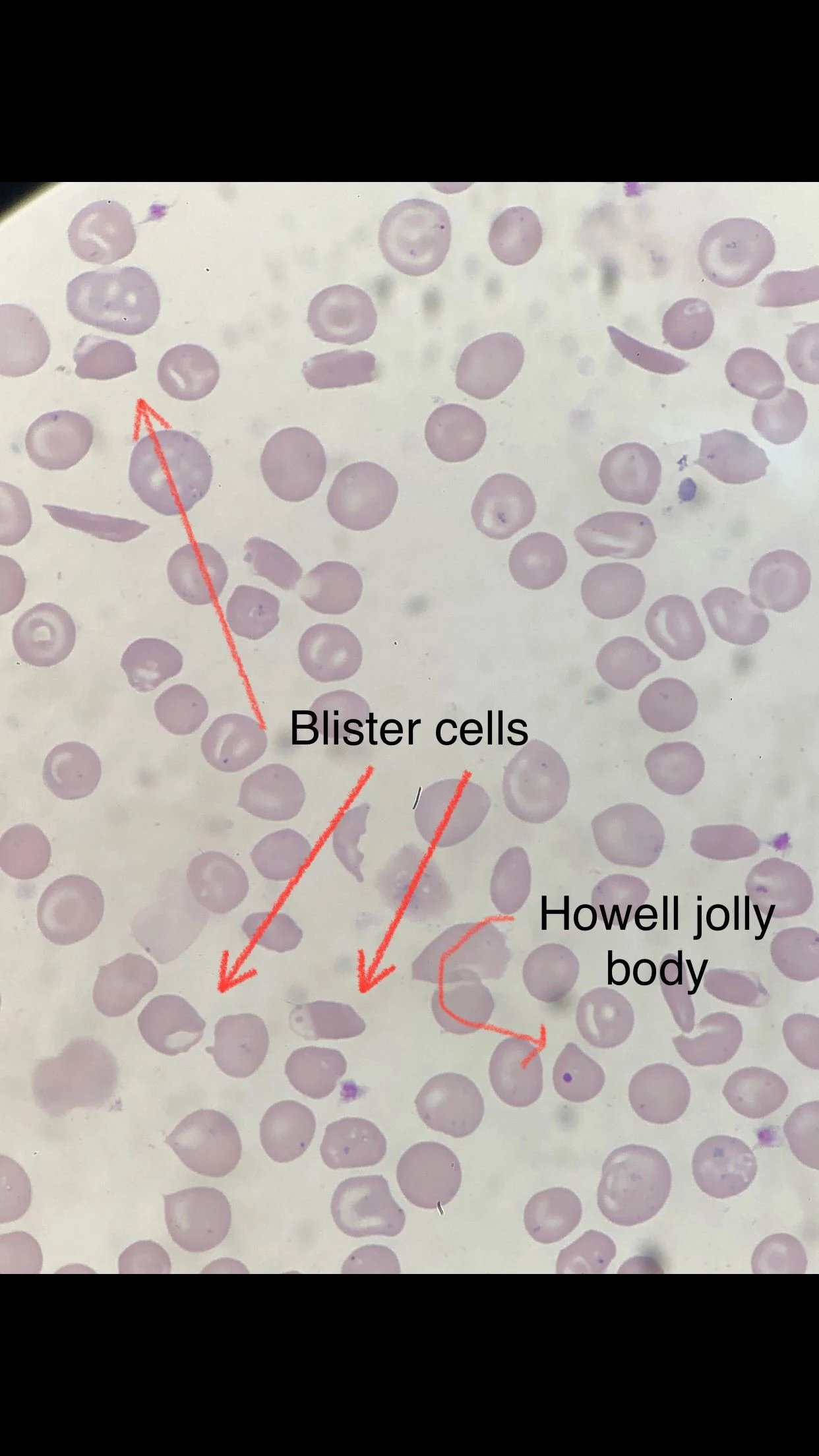
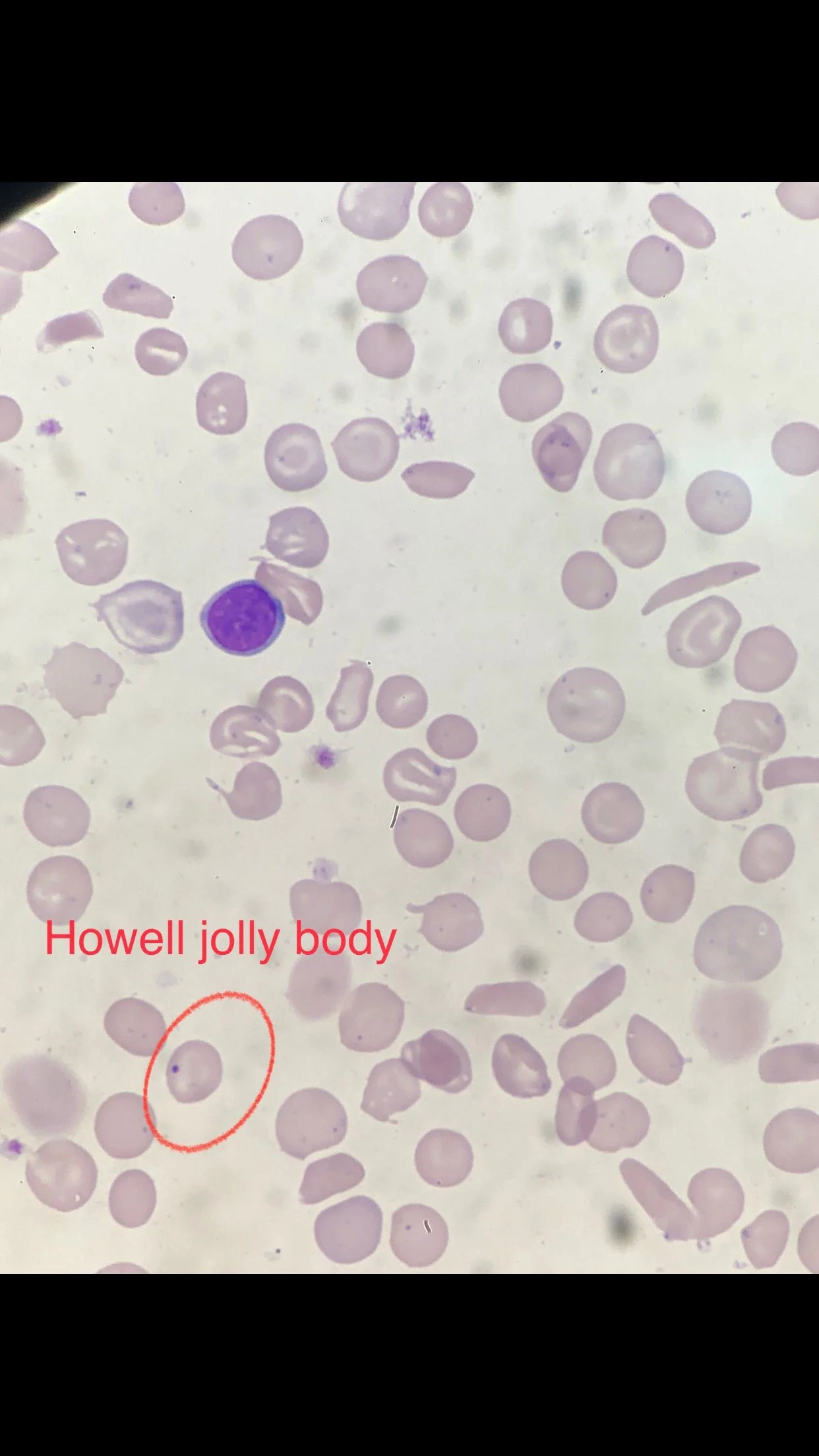
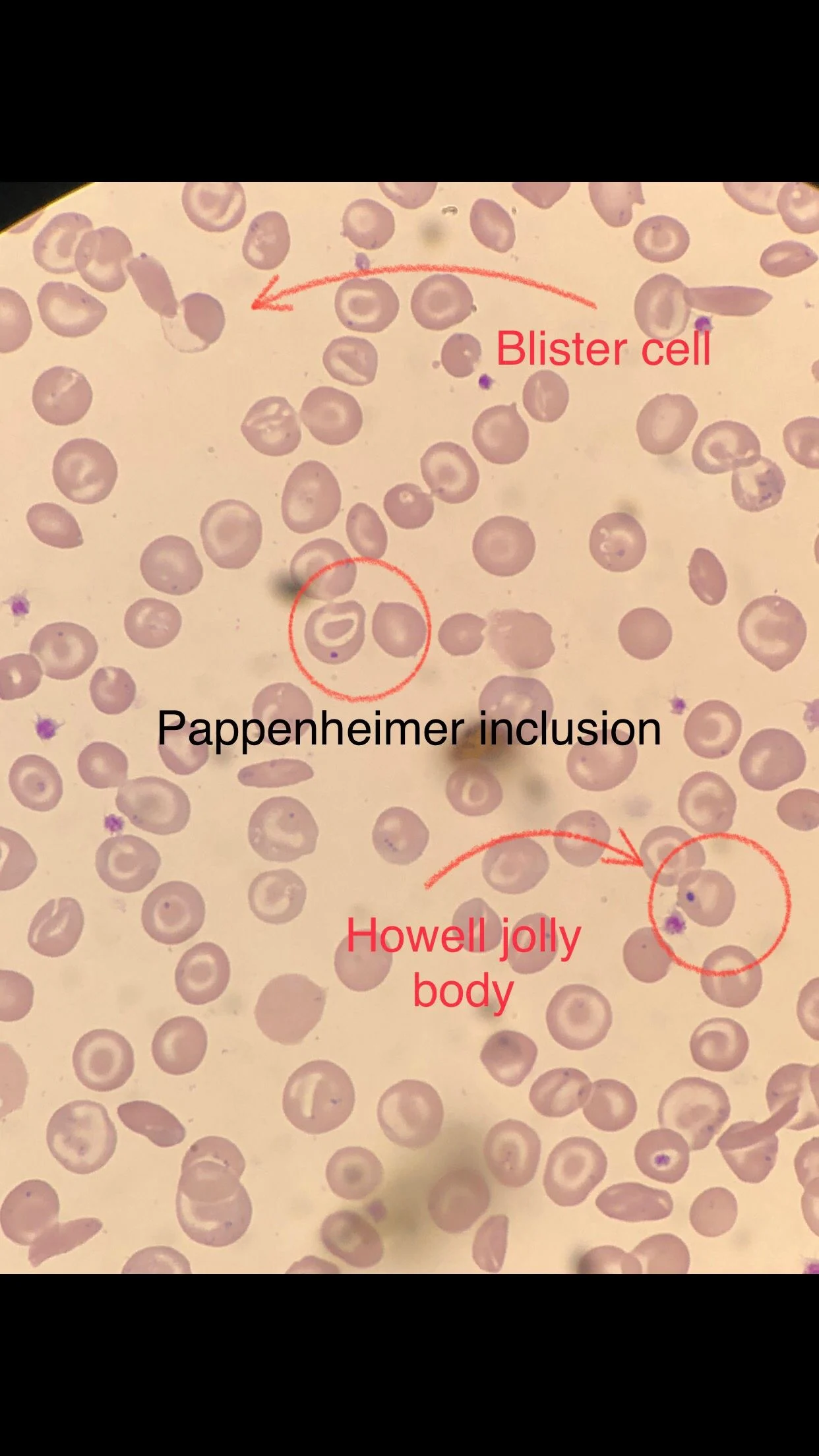
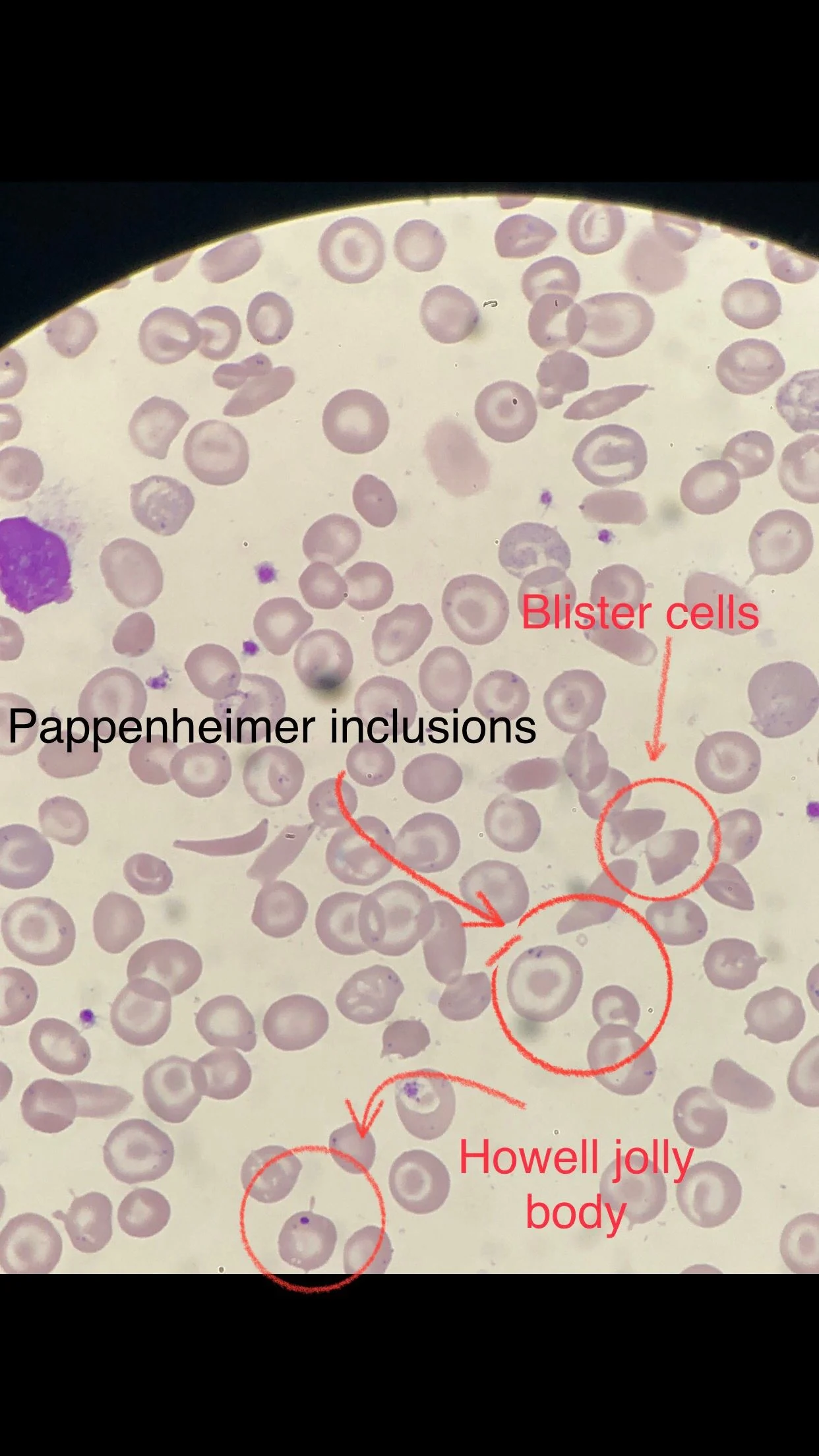
Many sickle cells. Target cells. Spherocytes. RBC fragments. Polychromasia. Pappenheimer inclusions. Howell jolly bodies. ovalocytes. but we also have bite cells and blister cells very clear!
So we did G6PD test and he was deficiency also.
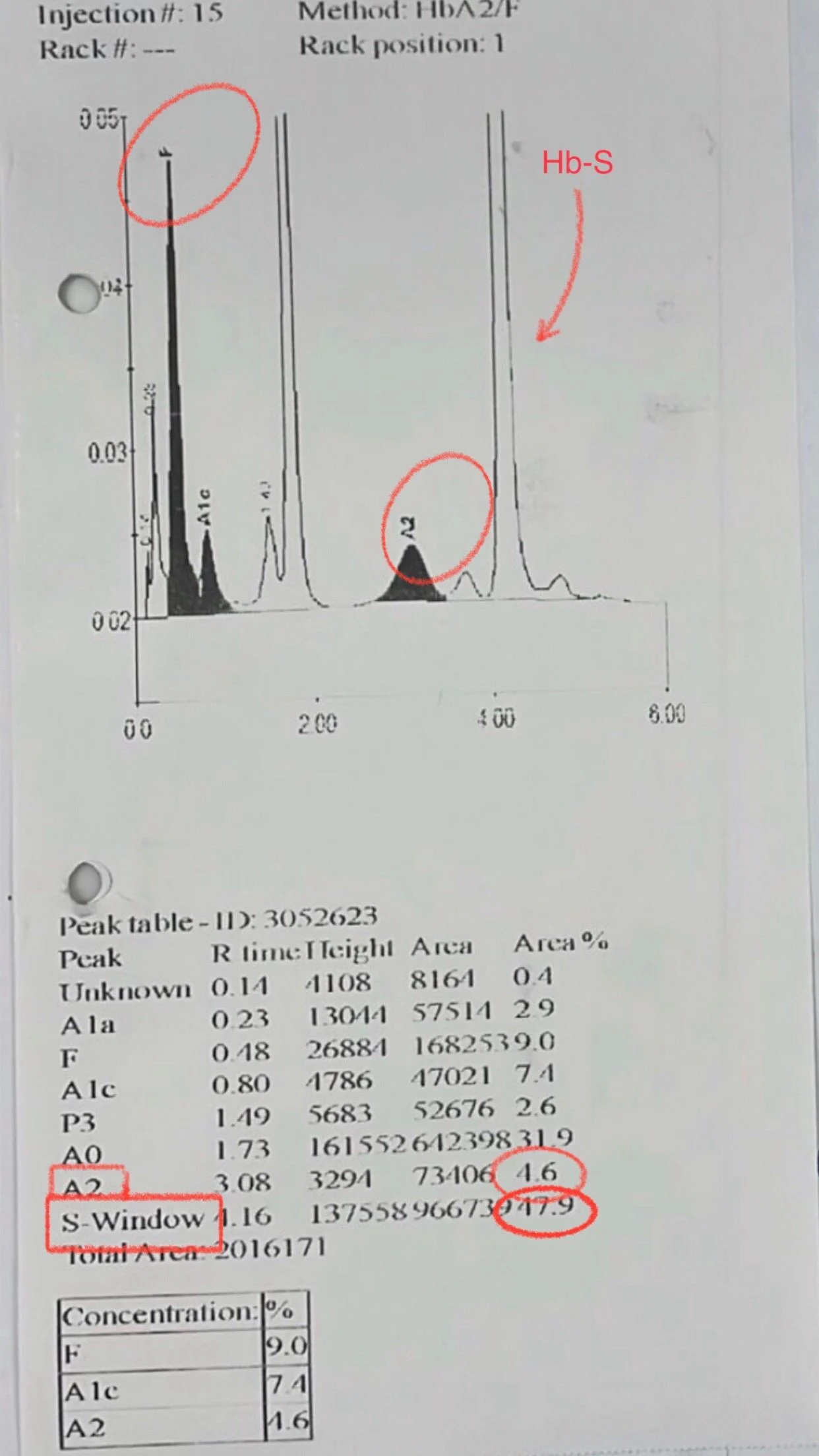
Hemoglobin electrophoresis done after that and we can see Hb-S after transfusion went down to 47.9 %.
But Hb-A2 was high 4.6 %. meaning he has beta thalassemia trait. so this poor young man has sickle cell disease. beta thalassemia trait (sickle beta Thal) and G6PD deficiency.
A young man with the combination of sickle cell disease (SCD), beta-thalassemia trait (𝛽 – thal trait), and G6PD deficiency presents a complex hematological scenario.
This combination involves two distinct inherited disorders causing hemolytic anemia – SCD and G6PD deficiency – plus a trait that modifies red blood cell (RBC) production.
The interaction of these conditions can influence the severity of the clinical course of SCD, potentially increasing the frequency of hemolytic crises.
The mild Leukocytosis with Neutrophilia might not be due to any infection.
Sickle beta thalassemia (HbS/β-thalassemia) can cause high white blood cell (WBC) counts and neutrophilia (elevated neutrophils) even in the absence of an active infection.
Now alhamduleallah he is in stable status and they explained to him his real case so he can take care for the complex status he has. he still young. wish the best and to be safe in his future and God bless you all.”
 Badran: Navigating the Clinical Complexity of Combined Red Cell Disorders 1")
Stay updated with all scientific advances with Hemostasis Today.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
-
Jul 26, 2026, 08:51Dayita Banerjee: Presenting New Insights in Fibrinolysis Research at ISTH 2026
-
Jul 26, 2026, 08:39Roshni Kulkarni: The Human Face of Postpartum Hemorrhage
-
Jul 26, 2026, 08:14Atahar imam Hasan: Universal Artificial Blood and the Future of Trauma Care
-
Jul 26, 2026, 07:40Maha Othman: Leading Scientific Collaboration Across Hemostasis at ISTH 2026
-
Jul 26, 2026, 07:20Pablo Corral: Why Should Dyslipidemia Be Any Different?
-
Jul 26, 2026, 06:46Marios Georgakis: Largest GWAS of Transient Ischemic Attacks Reveals New Genetic Insights into Stroke Risk
-
Jul 26, 2026, 06:40Aaron Iding: From Virtual Meetings to Meaningful Connections at ISTH 2026
-
Jul 26, 2026, 06:36How Stroke Services Have Evolved Over the Past 8 Years in Kazakhstan – International Journal of Stroke
-
Jul 26, 2026, 06:31Leonardo Roever: Non-LDL Biomarkers and The Potential Future Applications for 2ry Stroke Prevention